dNdS-Fun
A tool for detecting selection signatures of both coding and noncoding somatic mutations in cancer genomes
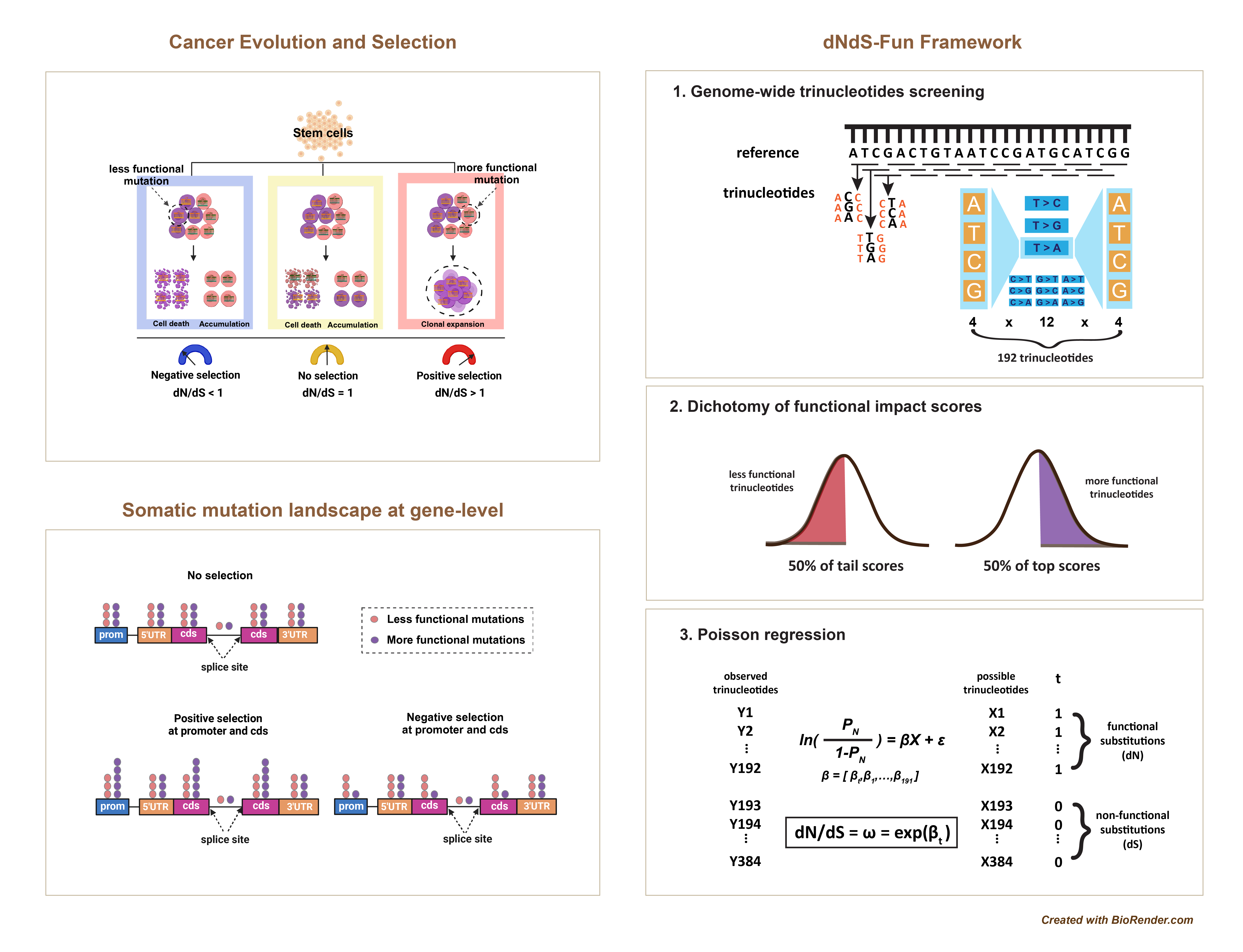
dNdS-Fun is a generalized framework that extends the classical dN/dS methodology, specifically dNdScv, to detect and quantify selection signatures on both coding and noncoding somatic mutations in cancer genomes. By integrating genome-wide functional impact scores, dNdS-Fun allows for the identification of positive and negative selection of both coding and noncoding mutations at global (genome-wide) and local (gene or element-specific) scales.
Key Features
- Functional Impact Scores Integration: Utilizes genome-wide functional impact scores (e.g., CADD) to assess the potential functional importance of mutations across the entire genome.
- Mutation Grouping: Classifies genomic sites into two groups based on functional impact scores:
- More Functional Group: Sites with higher functional impact scores (analogous to nonsynonymous mutations).
- Less Functional Group: Sites with lower functional impact scores (analogous to synonymous mutations).
- Selection Metric (ω): the normalized ratio of observed mutations in the more functional group to the less functional group, adjusted for the number of possible sites and mutation rates. The ω ratio indicates the direction of selection and quantifies selection strength:
- ω > 1: Indicates positive selection.
- ω < 1: Indicates negative selection.
- ω = 1: Indicates no evidence of selection.
- Trinucleotide Context Correction: Following dNdScv, accounts for sequence context-dependent mutation rates by modelling mutations within a 192 trinucleotide framework (considering all possible substitutions in the context of one upstream and one downstream base).
- Global and Local Analysis:
- Global Analysis: Estimates ω within a functional category (e.g., coding sequences, promoters, splice sites, UTRs, introns, intergenic regions) across the entire genome.
- Local Analysis: Estimates ω for individual genes or genomic elements, allowing for fine-scale detection of selection signatures.
Statistical Modelling
- Negative Binomial Regression for Local Analysis: Models the observed mutation counts using a negative binomial distribution to account for overdispersion and varying mutation rates across genes or elements.
- Likelihood Ratio Test (LRT): Performs statistical testing to determine if the observed ω significantly deviates from neutrality (ω ≠ 1), indicating selection. P-values are derived from a chi-square distribution with one degree of freedom.
Usage
- Input Data Requirements:
- Somatic mutation data with genomic positions and functional impact scores.
- Adjustments for Mutation Rates:
- Corrects for mutation rate variability due to sequence context and regional differences.
- Incorporates trinucleotide mutation models and gene-specific covariates.
- Applicability:
- Suitable for WGS or WES data.
- Can be applied to both coding and noncoding regions.
- Detects both positive and negative selection signatures.
Advantages
- Extension to Noncoding Regions: Expands traditional dN/dS analysis beyond coding regions by using functional impact scores to evaluate noncoding mutations.
- Unified Framework: Provides a consistent method to detect selection across the entire genome, facilitating comprehensive analysis.
- Robust to Mutation Rate Variability: Corrects for context-dependent mutation rates and overdispersion, ensuring accurate estimation of selection pressures.
- Versatility: Capable of analyzing different functional categories and accommodating various functional impact scoring systems, and compatible with both GRCh37 and GRCh38 genome builds.
Validation
- Simulations: Demonstrated reliability and accuracy in estimating selection parameters under neutral and selected scenarios through extensive simulations.
- Benchmarking: Outperformed existing driver discovery methods in precision and recall when applied to both simulated and real cancer genomic data.
- Reproducibility: Showed consistent results across different datasets (e.g., TCGA, PCAWG, 100kGP) and sequencing platforms (WES and WGS).
Implementation
- Software Availability: dNdS-Fun is available as an open-source software tool on GitHub. We have also developed a web-based platform (https://yanglab.westlake.edu.cn/dNdS-Fun/) for online data analysis.
- Programming Language: Implemented in R, leveraging existing statistical packages for regression modelling and statistical testing.
- User Documentation: Comprehensive documentation, including installation instructions, usage examples, and parameter explanations, is provided to guide users.
- Customization: Users can adjust parameters such as functional impact score thresholds and include additional covariates to tailor the analysis to specific datasets.
Conclusion
dNdS-Fun represents a significant advancement in detecting selection in cancer genomes by enabling analysis of both coding and noncoding regions within a unified framework. By integrating functional impact scores and accounting for mutation rate variability, dNdS-Fun provides a powerful tool for identifying driver genes and elements under selection, offering valuable insights into tumorigenesis and potential therapeutic targets.